Development notes
During the development of this workflow, we have made a number of decisions and ran into unexpected situations when using certain tools. This document describes the rationale of developing the workflow in its current form and sheds light on why we use the tools and parameters we use.
Downloading databases
(Rationale for including in the Snakefile or providing separate scripts
that need to be run before running the workflow with snakemake.
This item needs to be updated!)
Databases are downloaded for:
-
ATB input genomes (script
bin/prepare_genomes.sh) -
Multilocus sequence typing (Snakemake rule
download_mlst_database) -
PADLOC anti-phage defence system screening (Snakemake rule
download_padloc_database) -
geNomad plasmid/phage predictions (not included, undocumented)
-
SpacePHARER CRISPR target prediction (script
download_spacepharer_database.sh)
Use of GNU parallel over Snakemake
Snakemake is very good at running processes in parallel and making efficient use of computational resources. However, the file-based nature combined with the necessity to generate a directed acyclic graph (DAG) before running the actual workflow may lead to excessive waiting times when you have tens of thousands of input files. Therefore, we decided to use the batches provided by AllTheBacteria as input units and use a separate program, GNU parallel, to handle the files within these batches. This solution is taken from this online post by John van Dam: https://stackoverflow.com/a/55007872. As described there, this makes the whole DAG simpler, and the processing faster, at the cost of losing checks by Snakemake to see if all expected output files are generated.
Processing batches as concatenated fasta files
As an alternative method, the fasta files for each batch may be concatenated in one long file with multiple genomes and processed at once. Tools that are adapted to analysing metagenomes can process separate fasta files as well as such a concatenated file. Besides, it may save overhead time while still taking advantage of multiple CPUs if the program is multithreaded.
We found that CCTyper will happily run on concatenated input files, and so will Jaeger. CRISPRidentify can take an input folder with many fasta files inside, and PADLOC would not finish when running on an input file containing thousands of genomes.
For CCTyper and Jaeger, we did a little benchmark of 4 batches to test for speed differences between the 'concatenate' and 'parallel' methods.
CCTyper using the parallel method used 1:40:00h to finish a small batch,
and up to 23:50:00h for large batches. With the input files concatenated
into one fasta per batch, the times were 39:00m and 12:07:00h, respectively.
That means concatenating input resulted in 2.56 times faster analysis
of the small batch and 1.97 times faster for the large batch.
We estimate that this method speeds up the analysis by 2-3 fold.
(We used the --prodigal meta setting to accomodate for having
different genomes in the same file.)
For CCTyper runtimes for all Campylobacter batches in AllTheBacteria,
see this figure:\
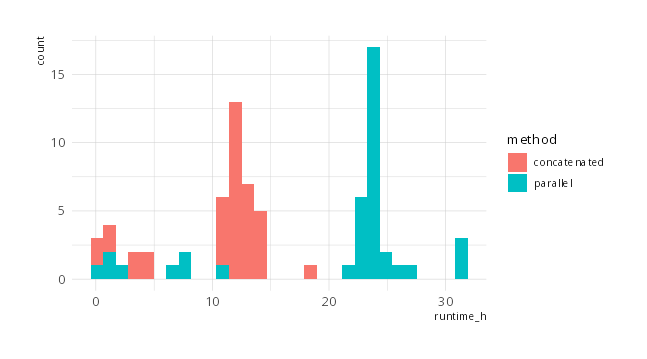
Jaeger used 17:22m and 7:12:00h on the small and large batch, respectively, using the parallel method. When concatenating the input, the times were 6:45m and 1:53:00h. That translates to a speedup of 2.57 - 4.63 times, or roughly 2.5-4.5 fold.
Besides, concatenating all input files also makes it easier to collect outputs from the whole dataset. Together with the observed speedups, we have decided to apply this 'concatenation method' as much as possible.
For details, see my minibenchmark notes.
Memory use for concatenated batches
While running CCTyper on concatenated input files made the analysis faster, memory use also increased which led to problems when running multiple CCTyper processed simultaneously. Especially with the settings for 'extra sensitive' CRISPR array detection (see commit 88e8fdb and description in version 0.3.1), memory consumption per process would be >100GB, making RAM a bottleneck.
To circumvent this huge RAM consumption, the rule for CCTyper has been changed to cut Campylobacter batches in chunks of up to 50 million nucleotides each. This decreased total RAM use to more manageable number of around 40GB/job. See figure below.
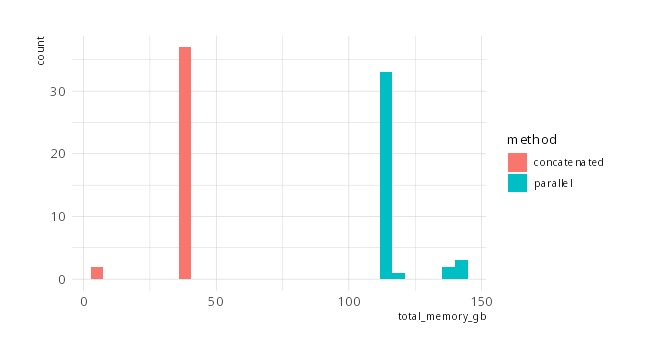
Results may depend on batch processing method
When analysing batches of ATB genomes, we found two ways to process genomes in parallel and speed up the whole process. One is to use GNU parallel to read multiple input genomes at the same time. The other is to concatenate all input files and use multiple threads for processing the whole batch simultaneously. We hypothesised this would generate the exact same output, but found that running CCTyper in these two different ways yielded slightly different output files. We found that the file-by-file approach in one particular batch would return 7 hits that were not found from the concatenated file, while the concatenated approach yielded 1 hit that was not found in the separate genomes. We did not pursue this discrepancy further, but keep in mind that minor differences may occur between reruns of the same analysis on identical inputs.
Using other inputs than ATB
This workflow was designed to work with bacterial genomes from
AllTheBacteria (ATB)
as input and has provided scripts to automatically download genomes
of the bacterial species of interest.
However, the processing steps should work equally well on other genomes in
FASTA format. Currently, the easiest way to 'trick' the workflow into using
custom input is to use the expected input folder structure:
resources/ATB/assemblies/
and create a folder there with your own genome sequences.
The name of the directory has to start with atb.assembly.,
and the fasta files should have the '.fa' extension, so for example:
mkdir -p resources/ATB/assemblies/atb.assembly.my_test
mv /path/to/genomes/*.fa resources/ATB/assemblies/atb.assembly.my_test/
If you have genome sequences in the directory /path/to/genomes/
with a '.fa' file extension, this will move them into the batch folder "my_test",
and that makes CRISPRscape see your genomes as input.
Sample names in fasta files
Note that all fasta files in the batch will be concatenated into
one long fasta file. To separate them back into their corresponding
samples, you need to have the sample names in the sequence IDs of
the fasta files. For example, in ATB fasta IDs are like:
SAMN30577462.contig00001 len=228074 cov=48.3 corr=0 origname=NODE_1_length_228074_cov_48.273485_pilon sw=shovill-spades/1.1.0 date=20230630
Therefore, CRISPRscape works best if you follow this
'[sample].[contig]' naming convention in the fasta files.
(Everything after the space is only for your own convenience and
is not explicitly used in the workflow.)
Metadata for dereplication
The workflow tries to automatically dereplicate input genomes, for
which it uses the ATB metadata file checkm2.tsv.gz. If this file
is not present, the whole dereplication process will fail, and you
will be presented errors in your terminal window! This may safely
be ignored if duplicates are not a concern, or you may remove the
line "results/dereplication_table.tsv", from the Snakefile
to disable automated dereplication.
Modified CCTyper installation
We found that the default conda-based installation of CRISPRCasTyper would
create a non-functional program. We have reported this on
GitHub and provide
a modified software environment configuration file (envs/cctyper.yaml)
that we found to work.
For example, CCTyper uses import pkg_resources, which is a Python
setuptools library that has been removed since version 82:
setuptools changelog.
As a simple workaround, we include version 81.0.0.
Trying to be economical with disk space use
Downloading thousands of genomes from ATB and unpacking them into plain FASTA files needs quite a lot of space. We considered gzipping each fasta file separately to save disk space while still allowing most tools to work with them. However, we found CCTyper would not take gzipped input files, so we left the input files uncompressed.
Unexpected behaviour of tools and databases
Spacepharer
We found that Spacepharer wants to run within the same folder that you designate the tmpfolder and where the created its databases. This needs to be taken into account when writing the commands for the workflow.
Also, following the easy-predict workflow is not recommended as created
.fasta files are inconsistently recognized as actual fasta files.
Spacepharer databases are best created using the example names
"querysetDB" and "targetsetDB" as other names such as "spacersetDB"
causes weird errors.
Phagescope
The Phagescope database has to be downloaded in full, no selections can be made beforehand. Therefore, the download is pretty large, while the parts that are needed may be modest in size and can be filtered after downloading.
CRISPRidentify
The installation of CRISPRidentify was, unfortunately, not as straightforward as running
or similar. We found that the YAML file provided on the CRISPRidentify GitHub page cannot be solved using the recommended 'strict channel priority' setting. We could only get it to work with the flexible or disabled channel priority. We have adapted to YAML so that it can be solved using strict priority mode, but with this method two modules are disabled: strand and cas prediction (CRISRPcasIdentifier).
CCTyper
CCTyper assumes its output directory does not yet exist, while Snakemake will automatically prepare directories of the rules it is running. To solve this conflict, the rule running CCTyper has to start with something like: